ForceConstants
The ForceConstants object contains the force constants, supercell, and
crystal structure information required to calculate phonon frequencies and
eigenvectors at any arbitrary q via Fourier interpolation.
For faster, but less accurate linear interpolation with the Brille library, see Brille Interpolator.
Force Constants Format
Description
The ForceConstants.force_constants attribute contains the force constants matrix
This describes the change in total energy when atom \(\kappa\) in unit cell \(0\) is displaced in direction \(\alpha\) and atom \(\kappa\prime\) in unit cell \(l\) is displaced in direction \(\alpha\prime\). In Euphonic the force constants are stored in ‘compact’ form in a matrix of \(N(3n)^2\) elements, where \(N\) is the number of cells in the supercell, and \(n\) is the number of atoms in the unit cell. This form can be expanded as needed to a ‘full’ \((3Nn)^2\)-element matrix of supercell atom pairs by mapping to an equivalent vector from unit cell 0.
Shape and Indexing
The force constants matrix has shape (N, 3*n, 3*n), where N is the number of unit cells in the supercell, and n is the number of atoms in the unit cell.
The force constants index [l, a, b], where a = 3*kappa + alpha and b = 3*kappa_prime + alpha_prime,
describes the change in energy when atom kappa in unit cell 0 is displaced in direction alpha and atom kappa_prime in unit cell l is displaced in direction alpha_prime.
For example, ForceConstants.force_constants[5, 8, 1] is the change in energy when atom
2 in unit cell 0 is displaced in the z direction
and atom 0 in unit cell 5 is displaced in the y direction.
Phase Convention
To calculate phonon frequencies and eigenvectors, the dynamical matrix at wavevector \(\mathbf{q}\) must be calculated. This is calculated by taking a Fourier transform of the force constants matrix
The \(\mathbf{q}\cdot \mathbf{R}_l\) component is the phase, and in Euphonic’s convention \(\mathbf{R}_l\) is defined as the vector from the origin to the origin coordinate of the \(l^\textrm{th}\) unit cell in the supercell.
For this reason the cell origin coordinates are required as input to create a ForceConstants object.
For force constants from programs that use the same phase convention (such as CASTEP), this information is usually provided so is straightforward.
Force constants which come from programs where the phase is defined as \(\mathbf{q}\cdot \mathbf{R}_\kappa\) using the coordinate of each atom (such as Phonopy) are likely to need to be reshaped to be compatible with Euphonic, and the cell origins are not usually provided. If the unit cell and supercell are commensurate (i.e. the supercell matrix is diagonal), calculation of the cell origins and reshaping the force constants matrix should be trivial. However, if the unit and supercell are incommensurate there may be some reindexing required to get the force constants in the correct format. See the example below, which shows a 2D system with the following properties:
Cartesian unit cell vectors:
[1, 1], [1, -1]Fractional atom coordinates:
[0.8, 0.4], [0.2, 0.5]Supercell matrix:
[[1, -1], [1, 1]]
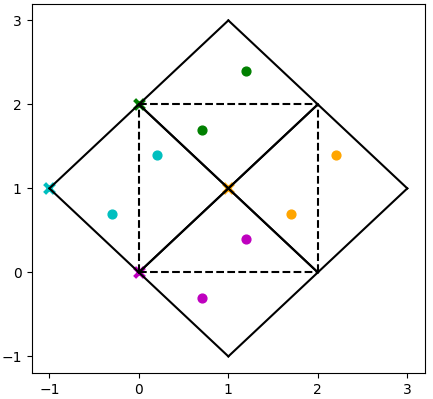
The solid line shows the unit cells, and the dashed line shows the supercell.
The atoms are coloured depending on the unit cell they belong to.
It can be seen that the supercell contains atoms from 4 different unit cells.
There would therefore be 4 different cell origins, shown by the corresponding coloured crosses.
However, there are actually only 2 unit cells in the supercell, and Euphonic requires that the number of cell origins is the same as the number of unit cells.
Using equivalent vectors in adjacent supercells, the 4 atoms in the supercell can be assigned to just 2 cell origins.
For example, if we decided to use the origins of the magenta cell ([0, 0]) and orange cell ([1, 1]), the blue and green atoms would have to be indexed into these 2 cell origins.
The way to do this can be seen in the example below:
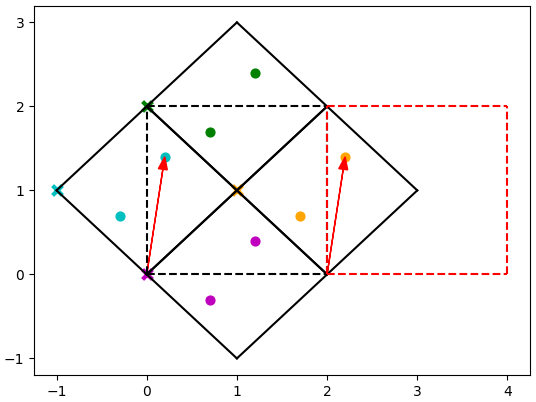
The vector from [0, 0] to the blue atom is the same as the vector from [2, 0] to an atom in the orange cell in the adjacent supercell (shown by the red dashed line).
The blue atom can therefore be indexed into the orange cell.
Similarly the green atom can be indexed into the magenta cell, so only 2 cell origins are required.
This is a simple example, but the same applies to more realistic 3D structures.
This transformation is done automatically when reading from Phonopy. If reading force constants from another program, there is a function to help with this transformation, see Reading From Other Programs.
Dipole-dipole Force Constants
For materials without dipole-dipole interactions (non-zero Born effective charges) force constants typically decay as \(1/r^{5-7}\), but if dipole-dipole interactions are present, they only decay at \(1/r^3\). It is often not computationally practical to compute the force constants at such a distance. Fortunately there is an analytical expression for these dipole interactions, which can be applied to the dynamical matrix to produce the correct phonon frequencies and displacements. However, to avoid double counting some of the dipole-dipole interactions, the force constants matrix from which the dynamical matrix is calculated must not already include the short-ranged part of the dipole interactions. This is represented in equation 78 in Gonze & Lee, 1997:
Where \(C\) is the ‘total’ force constants matrix containing both short-ranged interactions and long-ranged dipole interactions,
\(C^{SR}\) is the force constants matrix containing only the short-ranged interactions (i.e. no dipole interactions),
and \(C^{DD}\) is the force constants matrix containing only the long-ranged dipole interactions,
and all matrices have the same shape and indexing and contain interactions to the same cut-off distance.
To correctly apply the correction to the resulting dynamical matrix, \(C^{SR}\) must be used, which is what Euphonic requires.
Some codes (e.g. Phonopy) output \(C\) so must be converted.
This conversion is done automatically when reading from Phonopy, but if reading force constants from another program, there is a class method
ForceConstants.from_total_fc_with_dipole
which can do this transformation. An example of this is shown in Reading From Other Programs
Reading From CASTEP
The force constants matrix and other required information can be read from a
.castep_bin or .check file with
ForceConstants.from_castep:
from euphonic import ForceConstants
filename = 'quartz/quartz.castep_bin'
fc = ForceConstants.from_castep(filename)
By default CASTEP may not write the force constants, if you receive an error
saying the force constants could not be read, in the .param file ensure a
PHONON_FINE_METHOD has been chosen e.g. PHONON_FINE_METHOD: interpolate,
and set PHONON_WRITE_FORCE_CONSTANTS: true, then rerun CASTEP to trigger the
force constants to be written.
Reading From Phonopy
When using Phonopy with Euphonic, it is recommended that all the required data
(force constants, crystal structure, born charges if applicable) be collected
in a single phonopy.yaml file. This can be done by running Phonopy with the
--include-all flag or with INCLUDE_ALL = .TRUE.
(phonopy >= 2.5.0 only).
Required information is read from Phonopy output files using
ForceConstants.from_phonopy.
A path keyword argument can be supplied (if the files are in another
directory), and by default phonopy.yaml is read, but the filename can be
changed with the summary_name keyword argument:
from euphonic import ForceConstants
fc = ForceConstants.from_phonopy(path='NaCl',
summary_name='phonopy_nacl.yaml')
If you are using an older version of Phonopy, the force constants and born
charges can also be read from Phonopy plaintext or hdf5 files by specifying the
fc_name and born_name keyword arguments:
from euphonic import ForceConstants
fc = ForceConstants.from_phonopy(path='NaCl',
summary_name='phonopy_nacl.yaml',
fc_name='force_constants.hdf5',
born_name='BORN_nacl')
Reading From Other Programs
A Euphonic ForceConstants object can be created from arbitrary force constants and crystal information if they can be provided as Numpy arrays.
If the force constants are in the same form and shape as descibed in the Force Constants Format section, they can simply be used in the ForceConstants object constructor.
The crystal information must be provided as a Crystal object, which can similarly be created using the class constructor.
Some information must be provided as a pint Quantity with both a magnitude and a unit (see Units).
For more details on the shape of each array, see the docstrings for each object.
An example is shown below, assuming that the inputs are being loaded from existing .npy files:
import numpy as np
from euphonic import ureg, Crystal, ForceConstants
# Load arrays from files and apply any required units
cell_vectors = np.load('cell_vectors.npy')*ureg('angstrom')
atom_r = np.load('atom_r.npy')
atom_type = np.load('atom_type.npy')
atom_mass = np.load('atom_mass.npy')*ureg('amu')
force_constants = np.load('force_constants.npy')*ureg('meV/(angstrom**2)')
sc_matrix = np.load('sc_matrix.npy')
cell_origins = np.load('cell_origins.npy')
# Create a Crystal object
crystal = Crystal(cell_vectors, atom_r, atom_type, atom_mass)
# Create a ForceConstants object, using the Crystal object
fc = ForceConstants(crystal, force_constants, sc_matrix, cell_origins)
Changing Phase Convention
If, as described in the Force Constants Format section, the source program uses the atomic coordinate phase convention, there may be some re-indexing required to get the force constants in the correct shape and form.
There is a helper function euphonic.util.convert_fc_phases
which will convert a force constants of shape (n, N*n, 3, 3), to the shape required by Euphonic (N, 3*n, 3*n),
will do any re-indexing required, and will return the appropriate cell origins, even if the supercell matrix is non-diagonal.
All that is required are the coordinates of the atoms in the unit cell and supercell respectively,
the supercell matrix, and information on how atoms in the unit cell index into the supercell and vice versa.
For more details see the function docstring. An example is below:
import numpy as np
from euphonic import ureg, Crystal, ForceConstants
from euphonic.util import convert_fc_phases
# Load arrays from files and apply any required units
cell_vectors = np.load('cell_vectors.npy')*ureg('angstrom')
atom_r = np.load('atom_r.npy')
atom_type = np.load('atom_type.npy')
atom_mass = np.load('atom_mass.npy')*ureg('amu')
sc_matrix = np.load('sc_matrix.npy')
# Load arrays from files required for convert_fc_phases
force_constants_atomic_phase = np.load('force_constants_atomic_phase.npy')
supercell_atom_r = np.load('supercell_atom_r.npy')
unit_to_supercell_atom_idx = np.load('unit_to_supercell_atom_idx.npy')
super_to_unit_cell_atom_idx = np.load('super_to_unit_cell_atom_idx.npy')
# Convert force constants to Euphonic shape and convention
force_constants, cell_origins = convert_fc_phases(
force_constants_atomic_phase,
atom_r,
supercell_atom_r,
unit_to_supercell_atom_idx,
super_to_unit_cell_atom_idx,
sc_matrix)
# Apply units to force constants
force_constants = force_constants*ureg('meV/(angstrom**2)')
# Create a Crystal object
crystal = Crystal(cell_vectors, atom_r, atom_type, atom_mass)
# Create a ForceConstants object, using the Crystal object
fc = ForceConstants(crystal, force_constants, sc_matrix, cell_origins)
Dipole-dipole Force Constants - Converting from Total to Short-ranged
If, as described in the Force Constants Format section, the force constants matrix contains the dipole interactions, these must be subtracted to produce the short-ranged force constants matrix before being used in Euphonic.
This can be done using the class method
ForceConstants.from_total_fc_with_dipole.
Note that the force constants must have a Euphonic-like shape before using this method. An example is below:
import numpy as np
from euphonic import ureg, Crystal, ForceConstants
# Load arrays from files and apply any required units
cell_vectors = np.load('cell_vectors.npy')*ureg('angstrom')
atom_r = np.load('atom_r.npy')
atom_type = np.load('atom_type.npy')
atom_mass = np.load('atom_mass.npy')*ureg('amu')
force_constants = np.load('force_constants.npy')*ureg('meV/(angstrom**2)')
sc_matrix = np.load('sc_matrix.npy')
cell_origins = np.load('cell_origins.npy')
born = np.load('born.npy')*ureg('e')
dielectric = np.load('dielectric.npy')*ureg('e**2/(bohr*hartree)')
# Create a Crystal object
crystal = Crystal(cell_vectors, atom_r, atom_type, atom_mass)
# Create a ForceConstants object from the total force constants
fc = ForceConstants.from_total_fc_with_dipole(
crystal, force_constants, sc_matrix, cell_origins, born, dielectric)
Calculating Phonon Frequencies and Eigenvectors
Phonon frequencies and eigenvectors are calculated using
ForceConstants.calculate_qpoint_phonon_modes
(see the docstring for algorithm details). A Numpy array of q-points of shape
(n_qpts, 3) must be provided, and a
QpointPhononModes object is returned. A
recommended q-point path for plotting bandstructures can be generated using
seekpath:
import seekpath
import numpy as np
from euphonic import ForceConstants
# Read quartz data from quartz.castep_bin
filename = 'quartz/quartz.castep_bin'
fc = ForceConstants.from_castep(filename)
# Generate a recommended q-point path using seekpath
cell = fc.crystal.to_spglib_cell()
qpts = seekpath.get_explicit_k_path(cell)["explicit_kpoints_rel"]
# Calculate frequencies/eigenvectors
phonons = fc.calculate_qpoint_phonon_modes(qpts, asr='reciprocal')
Calculating Phonon Frequencies Only
This uses the same algorithm as for calculating both the frequencies and
eigenvectors, only with lower memory requirements as the eigenvectors
are not stored. This is done using
ForceConstants.calculate_qpoint_frequencies
which returns a QpointFrequencies object.
Docstring
- class ForceConstants(crystal, force_constants, sc_matrix, cell_origins, born=None, dielectric=None)
A class to read and store the data required for a phonon interpolation calculation from model (e.g. CASTEP) output, and calculate phonon frequencies/eigenvectors at arbitrary q-points via Fourier interpolation
- Variables
crystal – Lattice and atom information
force_constants – Shape (n_cells_in_sc, 3*n_atoms, 3*n_atoms) float Quantity in energy/length**2 units. The force constants matrix
sc_matrix – Shape (3, 3) int ndarray. The transformation matrix to convert from the unit cell vectors to the supercell vectors
n_cells_in_sc – Number of cells in the supercell
cell_origins – Shape (n_cells_in_sc, 3) int ndarray. The origin coordinates of each unit cell within the supercell, in units of the unit cell vectors
born – Shape (n_atoms, 3, 3) float Quantity in charge units or None. The Born charges for each atom
dielectric – Shape (3, 3) float Quantity in charge**2/(length*energy) units or None. The dielectric permittivity tensor
- __init__(crystal, force_constants, sc_matrix, cell_origins, born=None, dielectric=None)
- Parameters
crystal (
Crystal) – Lattice and atom informationforce_constants (
Quantity) – Shape (n_cells_in_sc, 3*n_atoms, 3*n_atoms) float Quantity in energy/length**2 units. The force constants matrixsc_matrix (
ndarray) – Shape (3, 3) int ndarray. The transformation matrix to convert from the unit cell vectors to the supercell vectorscell_origins (
ndarray) – Shape (n_cells_in_sc, 3) int ndarray. The origin coordinates of each unit cell within the supercell, in units of the unit cell vectorsborn (
Optional[Quantity]) – Shape (n_atoms, 3, 3) float Quantity in charge units. The Born charges for each atomdielectric (
Optional[Quantity]) – Shape (3, 3) float Quantity in charge**2/(length*energy) units. The dielectric permittivity tensor
- calculate_qpoint_phonon_modes(qpts, weights=None, asr=None, dipole=True, dipole_parameter=1.0, splitting=True, insert_gamma=False, reduce_qpts=True, use_c=None, n_threads=None, return_mode_gradients=False)
Calculate phonon frequencies and eigenvectors at specified q-points from a force constants matrix via Fourier interpolation
- Parameters
qpts (
ndarray) – Shape (n_qpts, 3) float ndarray. The q-points to interpolate onto in reciprocal cell vector unitsweights (
Optional[ndarray]) – Shape (n_qpts,) float ndarray. The weight for each q-point. If not given, equal weights are appliedasr (
Optional[Literal[‘realspace’, ‘reciprocal’]]) – Which acoustic sum rule correction to apply. ‘realspace’ applies the correction to the force constant matrix in real space. ‘reciprocal’ applies the correction to the dynamical matrix at every q-pointdipole (
bool) – Whether to calculate the dipole tail correction to the dynamical matrix at each q-point using the Ewald sum, if the Born charges and dielectric permitivitty tensor are present.dipole_parameter (
float) – Changes the cutoff in real/reciprocal space for the dipole Ewald sum. A higher value uses more reciprocal terms. If tuned correctly this can result in performance improvements. See euphonic-optimise-dipole-parameter program for help on choosing a good dipole_parameter.splitting (
bool) – Whether to calculate the LO-TO splitting at the gamma points. Only applied if dipole is True and the Born charges and dielectric permitivitty tensor are present.insert_gamma (
bool) – If splitting is True, this will insert gamma points into qpts to store the extra split frequencies. Note this means that the length of qpts in the output QpointPhononModes object will not necessarily be the same as the input qpts. If qpts already contains double gamma points where you want split frequencies, leave this as False.reduce_qpts (
bool) – Whether to use periodicity to reduce all q-points and only calculate for unique q-points within the 1st BZ. This won’t change the output but could increase performance.use_c (
Optional[bool]) – Whether to use C instead of Python to calculate and diagonalise the dynamical matrix. By default this is None and will use the C extension if it is installed, and fall back to Python if not. If use_c=True, this will force use of the C extension and an error will be raised if it is not installed. If use_c=False, this will force use of Python, even if the C extension is installedn_threads (
Optional[int]) – The number of OpenMP threads to use when looping over q-points in C. By default this is None, in which case the environment variable EUPHONIC_NUM_THREADS will be used to determine number of threads, if this is not set then the value returned from multiprocessing.cpu_count() will be usedreturn_mode_gradients (
bool) – Whether to also return the vector mode gradients (in Cartesian coordinates). These can be converted to mode widths and used in adaptive broadening for DOS. For details on how these are calculated see the Notes section
- Return type
Union[QpointPhononModes,Tuple[QpointPhononModes,Quantity]]- Returns
qpoint_phonon_modes – A QpointPhononModes object containing the interpolated frequencies and eigenvectors at each q-point. Note that if there is LO-TO splitting, and insert_gamma=True, the number of input q-points may not be the same as in the output object
mode_gradients – Optional shape (n_qpts, n_branches, 3) float Quantity, the vector mode gradients dw/dq in Cartesian coordinates. Is only returned if return_mode_gradients is true
- Raises
ImportCError – If use_c=True but the C extension cannot be imported
Notes
Phonon Frequencies/Eigenvectors Calculation
Phonon frequencies/eigenvectors are calculated at any q-point by Fourier interpolation of a force constants matrix. The force constants matrix is defined as 1:
\[\Phi_{\alpha {\alpha}^\prime}^{\kappa {\kappa}^\prime}(0,l) = \frac{\partial^{2}E}{{\partial}u_{\kappa\alpha0}{\partial}u_{{{\kappa}^\prime}{{\alpha}^\prime}l}}\]where the unit cell containing the displaced atom is labelled \(0\) and \(l\) runs over unit cells in the crystal, \(\kappa\) runs over atoms in the unit cell, \(\alpha\) runs over the Cartesian directions, and \(u_{\kappa{\alpha}l}\) is the displacement of atom \(\kappa\) in cell \(l\) in direction \(\alpha\) from its equilibrium position. This can be used to calculate the dynamical matrix \(D_{\alpha {\alpha}^\prime}^{\kappa {\kappa}^\prime}\) at \(\mathbf{q}\):
\[D_{\alpha {\alpha^\prime}}^{\kappa {\kappa^\prime}}(\mathbf{q}) = \frac{1}{\sqrt{M_\kappa M_{\kappa^\prime}}} \sum_{l}\Phi_{\alpha {\alpha^\prime}}^{\kappa {\kappa^\prime}}\exp\left(-i\mathbf{q}\cdot \mathbf{R}_l\right)\]where \(M_\kappa\) is the mass of atom \(\kappa\), \(\mathbf{R}_l\) is the vector from the origin to the \(l^\textrm{th}\) unit cell. The eigenvalue equation for the dynamical matrix is then:
\[\sum_{\kappa^\prime \alpha^\prime}D_{\alpha {\alpha}^\prime}^{\kappa {\kappa}^\prime}(\mathbf{q}) {e}_{\mathbf{q}\nu{\kappa^\prime}{\alpha^\prime}} = {\omega}_{\mathbf{q}\nu}^2 {e}_{\mathbf{q}\nu\kappa\alpha}\]where the eigenvalues \({\omega}_{\mathbf{q}\nu}^2\) are the square of the phonon frequencies of mode \(\nu\) at \(\mathbf{q}\), and the eigenvectors \(\mathbf{e}_{\mathbf{q}\nu\kappa}\) are the phonon polarisation vectors at \(\mathbf{q}\) of mode \(\nu\) for atom \(\kappa\). The produced eigenvectors are normalised such that:
\[\sum_{\kappa}{|\mathbf{e}_{\mathbf{q}\nu\kappa}|^2} = 1\]In polar materials, there is an additional long-ranged correction to the force constants matrix (applied if dipole=True) and a non-analytical correction at the gamma point 2 (applied if splitting=True).
- 1
Dove, Introduction to Lattice Dynamics, Cambridge University Press, Cambridge, 1993, 83-87
- 2
Gonze, J. C. Charlier, D. C. Allan, M. P. Teter, Phys. Rev. B, 1994, 50, 13035-13038
Mode Gradients Calculation
The mode gradients can be used to calculate mode widths to be used in the adaptive broadening scheme 3 when computing a DOS - this broadens each mode contribution individually according to its mode width. The mode widths are proportional to the mode gradients and can be estimated using
euphonic.util.mode_gradients_to_widthsThe mode gradients \(\frac{d\omega_{\mathbf{q}\nu}}{d\mathbf{q}}\) at each \(\mathbf{q}\) are calculated at the same time as the phonon frequencies and eigenvectors as follows.
Firstly, the eigenvalue equation above can be written in matrix form as:
\[E(\mathbf{q})\Omega(\mathbf{q}) = D(\mathbf{q})E(\mathbf{q})\]\[\Omega(\mathbf{q}) = E^{-1}(\mathbf{q})D(\mathbf{q})E(\mathbf{q})\]Where \(\Omega(\mathbf{q})\) is the diagonal matrix of phonon frequencies squared \(\omega_{\mathbf{q}\nu}^{2}\) and \(E(\mathbf{q})\) is the matrix containing eigenvectors for all modes. \(\frac{d\omega_{\mathbf{q}\nu}}{d\mathbf{q}}\), can then be obtained by differentiating the above equation with respect to \(\mathbf{q}\) using the product rule:
\[\frac{d\Omega}{d\mathbf{q}} = 2\omega_{\mathbf{q}\nu}\frac{d\omega_{\mathbf{q}\nu}}{d\mathbf{q}}\delta_{\nu \nu^{\prime}}\]\[\frac{d\omega_{\mathbf{q}\nu}}{d\mathbf{q}} = \frac{1}{2\omega_{\mathbf{q}\nu}}{( \frac{d{E^{-1}}}{d\mathbf{q}}DE + E^{-1}\frac{dD}{d\mathbf{q}}E + E^{-1}D\frac{dE}{d\mathbf{q}})}\]Given that eigenvectors are normalised and orthogonal, an identity can be employed:
\[\frac{d}{d\lambda}E^{\prime}E = 0\]So terms involving the first derivative of the eigenvector matrix can be set to zero, resulting in:
\[\frac{d\omega_{\mathbf{q}\nu}}{d\mathbf{q}} = \frac{1}{2\omega_{\mathbf{q}\nu}}{(E^{-1}\frac{dD}{d\mathbf{q}}E)}\]\(\frac{dD}{d\mathbf{q}}\) can be obtained by differentiating the Fourier equation above:
\[\frac{dD_{\alpha {\alpha}^\prime}^{\kappa {\kappa}^\prime}}{d\mathbf{q}} = \frac{-i \mathbf{R}_l}{\sqrt{M_\kappa M_{\kappa ^\prime}}} \sum_{l}\Phi_{\alpha\alpha^\prime}^{\kappa\kappa^\prime}\exp\left(-i\mathbf{q}\cdot \mathbf{R}_l\right)\]- 3
Yates, X. Wang, D. Vanderbilt and I. Souza, Phys. Rev. B, 2007, 75, 195121
- calculate_qpoint_frequencies(qpts, weights=None, asr=None, dipole=True, dipole_parameter=1.0, splitting=True, insert_gamma=False, reduce_qpts=True, use_c=None, n_threads=None, return_mode_gradients=False)
Calculate phonon frequencies (without eigenvectors) at specified q-points. See ForceConstants.calculate_qpoint_phonon_modes for argument and algorithm details
- Return type
Union[QpointFrequencies,Tuple[QpointFrequencies,Quantity]]
- to_dict()
Convert to a dictionary. See ForceConstants.from_dict for details on keys/values
- Return type
Dict[str,Any]- Returns
dict
- to_json_file(filename)
Write to a JSON file. JSON fields are equivalent to ForceConstants.from_dict keys
- Parameters
filename (
str) – Name of the JSON file to write to- Return type
None
- classmethod from_dict(d)
Convert a dictionary to a ForceConstants object
- Parameters
d (
Dict[str,Any]) –A dictionary with the following keys/values:
’crystal’: dict, see Crystal.from_dict
’force_constants’: (n_cells_in_sc, 3*crystal.n_atoms, 3*crystal.n_atoms) float ndarray
’force_constants_unit’: str
’sc_matrix’: (3,3) int ndarray
’cell_origins’: (n_cells_in_sc, 3) int ndarray
There are also the following optional keys:
’born’: (3*crystal.n_atoms, 3, 3) float ndarray
’born_unit’: str
’dielectric’: (3, 3) float ndarray
’dielectric_unit’: str
- Return type
TypeVar(T, bound= ForceConstants)- Returns
forceconstants
- classmethod from_total_fc_with_dipole(crystal, force_constants, sc_matrix, cell_origins, born, dielectric)
Subtracts a dipole term from the input force constants matrix to convert the ‘total’ force constants matrix containing both dipole-dipole interactions and short-ranged interactions to one containing only short-ranged interactions, as defined in eq. 78 from 4 . This correction may need to be applied as Euphonic requires the short-ranged matrix, whereas some codes output the total matrix (e.g Phonopy, but note that the from_phonopy method will apply this correction automatically). If the force constants matrix is already short ranged or the material has zero Born effective charges or dielectric permittivity tensor this correction should not be applied.
- 4
Gonze and C. Lee., Phys. Rev. B, 1997, 55, 10355-10368
- Parameters
force_constants (
Quantity) – Shape (n_cells_in_sc, 3*n_atoms, 3*n_atoms) float Quantity in energy/length**2 units. The ‘total’ force constants matrix containing both dipole-dipole interactions and short-ranged interactions.crystal (
Crystal) – Lattice and atom informationsc_matrix (
ndarray) – Shape (3, 3) int ndarray. The transformation matrix to convert from the unit cell vectors to the supercell vectorscell_origins (
ndarray) – Shape (n_cells_in_sc, 3) int ndarray. The origin coordinates of each unit cell within the supercell, in units of the unit cell vectorsborn (
Quantity) – Shape (n_atoms, 3, 3) float Quantity in charge units. The Born charges for each atomdielectric (
Quantity) – Shape (3, 3) float Quantity in charge**2/(length*energy) units. The dielectric permittivity tensor
- Return type
TypeVar(T, bound= ForceConstants)- Returns
forceconstants
- classmethod from_json_file(filename)
Read from a JSON file. See ForceConstants.from_dict for required fields
- Parameters
filename (
str) – The file to read from- Return type
TypeVar(T, bound= ForceConstants)- Returns
forceconstants
- classmethod from_castep(filename)
Reads from a .castep_bin or .check file
- Parameters
filename (
str) – The path and name of the file to read- Return type
TypeVar(T, bound= ForceConstants)- Returns
forceconstants
- classmethod from_phonopy(path='.', summary_name='phonopy.yaml', born_name=None, fc_name='FORCE_CONSTANTS', fc_format=None)
Reads data from the phonopy summary file (default phonopy.yaml) and optionally born and force constants files. Only attempts to read from born or force constants files if these can’t be found in the summary file.
- Parameters
path (
str) – Path to directory containing the file(s)summary_name (
str) – Filename of phonopy summary file, default phonopy.yaml. By default any information (e.g. force constants) read from this file takes priorityborn_name (
Optional[str]) – Name of the Phonopy file containing born charges and dielectric tensor (by convention in Phonopy this would be called BORN). Is only read if Born charges can’t be found in the summary_name filefc_name (
str) – Name of file containing force constants. Is only read if force constants can’t be found in summary_namefc_format (
Optional[str]) – One of {‘phonopy’, ‘hdf5’}. Format of file containing force constants data, if it can’t be determined from the extension. FORCE_CONSTANTS is type ‘phonopy’
- Return type
TypeVar(T, bound= ForceConstants)- Returns
forceconstants